Se trata de desórdenes metabólicos de acumulación de glucógeno por deficiencia de enzimas muy importantes como la glucosa 6 fosfatasa y la α 1.4-glucosidasa lisosomal respectivamente..
Para la compresión más profunda sobre los procesos de glucogenogénesis (síntesis), glucogenolísis (degradación) ó gluconeogénesis (cliclo de Cori) les recomiendo Bioquímica de Herper ó de Werner.. en esta entrada solo nombraremos ciertos aspectos fundamentales a manera de introducción y nos enfocaremos en las manifestaciones patológica de las mismas..
El glucógeno es
conocido tambien como almidon animal, es un homo monosacárido, constituye la
principal fuente de almacenamiento de energía en la célula anímal y se almacena
principalmente en el higado (10-12%) y en el músculo (2%), se forma a partir de
miles de moléculas de glucosa conformado
por cadenas lienales unidas por enlace α-1,4- glucosídicos y ramificadas por
enlace α-1,6-glucosídicos.
Cumple funciones amortiguadoras, de vital importancia para regular los niveles de glucosa en la sangre de tal manera que se eviten niveles demasiado altos (hiperglicemia) que pueden acarrear ciertas patologías (diabetes mellitus) gracias a la presencia de insulina (producida por las céulas β del pancreas) la cual ayuda a la difusión facilitada de glucosa al interior de las células del organismos para su correcto funcionamiento y almacenamiento (hepatocitos)
Cumple funciones amortiguadoras, de vital importancia para regular los niveles de glucosa en la sangre de tal manera que se eviten niveles demasiado altos (hiperglicemia) que pueden acarrear ciertas patologías (diabetes mellitus) gracias a la presencia de insulina (producida por las céulas β del pancreas) la cual ayuda a la difusión facilitada de glucosa al interior de las células del organismos para su correcto funcionamiento y almacenamiento (hepatocitos)
Evitar también niveles muy bajos (hipoglicemia) que puedan ocasionar
diversos problemas (hipoxia celular y tisular y posteriormente su muerte), lo
cual se da gracias a la intervención del glucagón, (producido por las células α
del pancreas) el cual causa la degradación de glucógeno a glucosa, la cual es
liberada a la sangre.
Esta regulación glucémica se cumple por medio de dos procesos, la
glucogenogénesis y glucogenolisis.
SÍNTESIS - GLUCOGÉNESIS (Imagen 1)

DEGRADACIÓN -GLUCOGENÓLISIS (Imágen 2)

Ahora bien..
GLUCOGENOSIS
En la glucogenólisis la glucosa debe ser desfosforilada para abandonar la
célula, un transpostador de glucosa-6-fosfato
lleva la misma del citosol al retículo endoplasmático donde una enzima conocida
como glucosa-6-fosfatasa hidrolisa el ester del fosfato y la transforma en
glucosa libre, esta enzima se expresa únicamente en el hígado y en el riñon. Una
vez formada esta glucosa “libre” ésta puede abandonar el hígado y contribuir a
la tamponación de glucosa en la sangre, específicamente en el ayuno ó en la actividad
muscular.
Las deficiencias genéticas causan fallos en la función de las enzimas involucradas en la glucogenólisis y esto puede causar graves enfermedades de reserva de glucógeno, conocido como glucogenosis. La glucogenósis es una enfermedad hederitaria que se transmiten con carácter autosómico recesivo, y se da por el almacenamiento excesivo de este polisacárido.
Las deficiencias genéticas causan fallos en la función de las enzimas involucradas en la glucogenólisis y esto puede causar graves enfermedades de reserva de glucógeno, conocido como glucogenosis. La glucogenósis es una enfermedad hederitaria que se transmiten con carácter autosómico recesivo, y se da por el almacenamiento excesivo de este polisacárido.
TIPO
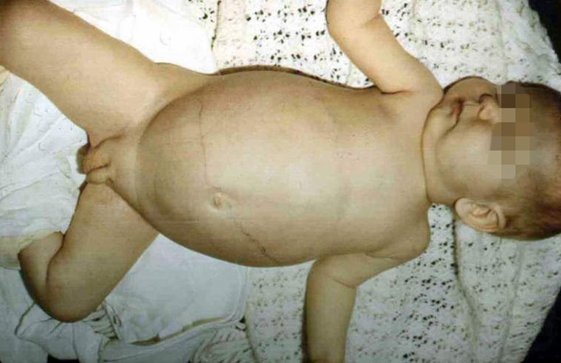
I - ENFERMEDAD DE VON - GIERKE
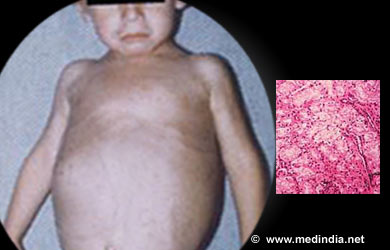
Ésta a su vez se divide en dos grupos: A y B
TIPO I-A
Ésta se produce por una deficiencia de la enzima glucosa-6-fosfatasa, la misma que desfosforila a la
glucosa-6-fosfato para su salida desde los hepatocitos hacia la sangre. Un
paciente con ésta patologia sufre una grave hipoglucemia y posteriormente se
produce una acumulación excesiva de glucógeno en el hígado y en riñon. Por ello
el hígado desvía el metabolismo de modo compensatorio hacia la glucólisis, lo
que conduce a una acumulación excesiva de piruvato y lactato provocando así una
acidosis metabólica (acumulación de iones H en el organismo)
TIPO I-B
En esta se observan sintomas similares a los de la glucegonisis de tipo I
–A, con defectos genéticos de los componentes de los transportadores de
glucosa-6-fosfato que se encargan de llevar la misma desde el citosol hacia el
retículo endoplasmático
En la glucogenosis de tipo I los
órganos afectados son el higado y el riñon, donde se observa que hay una
reserva de glucógeno excesiva, la principal expresión de esta anomalía son la
hepatomegalia, hipoglucemia y cetoacidosis.
PATOLOGÍAS ASOCIADAS
El fenotipo clínico es similar en todas ellas, aunque los pacientes con
el tipo I-B asocian neutropenia, constante o cíclica, cuya gravedad oscila
desde leve hasta la agranulocitosis y alteración de la función de los neutrófilos
que condicionan infecciones bacterianas recurrentes y úlceras bucales e
intestinales.
Los síntomas de presentación más frecuentes son:
Los síntomas de presentación más frecuentes son:
 ·
Hipoglucemia
severe,
·
Hipoglucemia
severe,
·
Hepatomegalia
·
Retraso del
desarrollo.
La hipoglucemia puede provocar crisis convulsivas que pueden comprometer
la vida del niño o su desarrollo psicomotor.
La hiperlactacidemia y la acidosis metabólica son hallazgos habituales que pueden causar polipnea y febrícula en ausencia de infección evidente. Cuando la hiperlactacidemia es permanente, en ayunas o desencadenada con el ayuno orienta hacia una glucogenosis I.
La hiperlactacidemia y la acidosis metabólica son hallazgos habituales que pueden causar polipnea y febrícula en ausencia de infección evidente. Cuando la hiperlactacidemia es permanente, en ayunas o desencadenada con el ayuno orienta hacia una glucogenosis I.
Ocasionalmente los pacientes con glucogenosis tipo I refieren diarrea y
en la tipo Ib se ha descrito una enfermedad inflamatoria intestinal, similar a
la enfermedad de Crohn
En la mayoría de las complicaciones puede existir una talla baja además de un retraso puberal, los enfermos no tratados presentan alrededor de la pubertad las siguientes complicaciones:
En la mayoría de las complicaciones puede existir una talla baja además de un retraso puberal, los enfermos no tratados presentan alrededor de la pubertad las siguientes complicaciones:
·
Hiperlipidemia.
·
Pancreatitis.
·
Mayor
susceptibilidad para el desarrollo de arteriosclerosis.
·
Afectación renal: nefromegalia bilateral y, de
forma progresiva, proteinuria, hipertensión arterial, litiasis renal,
hipofosforemia, nefrocalcinosis, glomeruloesclerosis y fibrosis intersticial
progresiva; todo ello puede ocasionar una insuficiencia renal, susceptible de
diálisis e incluso trasplante.
·
Hiperuricemia,
con episodios de gota en el adulto.
·
Acidosis
·
Infecciones
digestivas
DIAGNÓSTICO
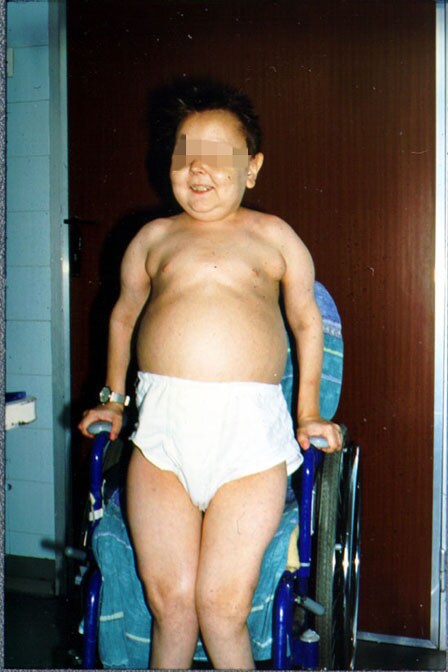
 El signo mas importante es la hepatomegalia (sin esplecnomegalia) nefromegalia asociada a esta, los pacientes
que padecen de glucogenosis tipo I poseen las facias redondeadas y obesidad
troncular, retraso de la edad ósea con ostepenia u osteoporosis
El signo mas importante es la hepatomegalia (sin esplecnomegalia) nefromegalia asociada a esta, los pacientes
que padecen de glucogenosis tipo I poseen las facias redondeadas y obesidad
troncular, retraso de la edad ósea con ostepenia u osteoporosis
El método más seguro de diagnóstico lo constituye la determinación del nivel
de actividad de la glucosa-6-fosfatasa en hígado. Las determinaciones de
glucemia y lactato en ayunas y las respuestas a la sobrecarga oral de glucosa y
al test del glucagón o las respuestas a la administración de galactosa o
fructosa.
TIPO
II – ENFERMEDAD POMPE

En esta ocasión la enzima definciente es la α-1,4-glucosidasa lisosomal por lo que el glucógeno aparece
almacenado en los lisosomas, todos los órganos están involucrados y presentan
una reserva de glucógeno excesiva, su principal sintomatología es la presencia
de fallos cardiorespiratorios como la insuficiencia cardíaca al acumularse en
el músculo cardíaco provocando asi una cardiomegalia y por lo general mueren
antes del segundo año de vida.
Se considera
como una enfermedad neuromuscular o una miopatía metabólica, las
manifestaciones clínicas primordiales del padecimiento son la debilidad y la
hipotonía, estas manifestaciones varían en función de la actividad residual de la enzima α-1,4-glucosidasa lisosomal, pero cuya historia natural
conduce a debilidad progresiva y muerte
debido a la acumulación de glucógeno.
Dependiendo el
fenotipo que presenta el paciente se ha llevado a clasificarla en dos tipos,
dependiendo de la edad de presentación, la de inicio temprano (infantil) y la de
inicio tardío (juvenil o del adulto),
PATOLOGÍAS
ASOCIADAS
Son características de la enfermeda:
 ·
Cardiomegalia,
·
Cardiomegalia,
·
Cardiomiopatía
·
Dificultad respiratoria,
·
Dificultad parala alimentación
·
Infecciones respiratorias repetidas
·
Falla para medrar.
·
Retraso global del desarrollo
·
Macroglosia y hepatomegalia (en algunos
pacientes)
DIAGNOSTICO
 En la forma más
grave que es la de inicio temprano se
caracteriza por:
En la forma más
grave que es la de inicio temprano se
caracteriza por:
·
Cardiomiopatía hipertrófica,
·
Hipotonía y debilidad muscular generalizada
·
Falla cardiorrespiratoria y muerte,
usualmente antes del año de edad.
La forma tardía
puede presentarse a cualquier edad, y se caracteriza por:
·
Disfunción musculoesquelética progresiva
(que puede involucrar a la función cardíaca)
Estas dos
anomalías se agraban progresivamente y la
edad de la muerte varía dependiendo de la velocidad de progresión de la
enfermedad, del grado de afección de los músculos respiratorios y de otras
comorbilidades
Para un diagnóstico
acertado no se debe basar únicamente en las patologias y aspecto fenotípico del
paciente, sino que se precisa de una biopsia muscular, que además de ser útil
para la cuantificación enzimática, permite conocer la cantidad de glucógeno
acumulado y su localización (intralisosomal y citosólica), lo cual ayuda a
establecer la existencia y gravedad de cada caso.
Como podemos constatar si buscamos un poco en la web, la gran
mayoría de los casos son encontrados por autopsia, puesto que la sintomatología
que este fallo metabólico (glucogenosis tipo II) dificulta en gran
medida un buen diagnóstico, por lo que los pacientes con este problema fallecen, y esto principalmente se da al tratamiento sintomático de estos problemas, más bien no se intenta o lograr ubicar la causa exacta de este padecimiento, por lo que los pacientes afectados experimentan una ligera mejoría y luego un gran decaimientoel cual los lleva a la muerte.
TRATAMIENTO
Al igual que las afecciones de carácter genético, no existe tratamiento que pueda curar estos desordenes metabólicos, sin embargo, existe un tratamiento sintomático, el cual no lograr correguir el problema pero puede brindar al paciente un estilo de vida mas llevadero.
ENFERMEDAD DE VON GIERKE (GLUCOGENOSIS TIPO I)
e
El objetivo del tratamiento es evitar la hipoglucemia. Como frecuentemente durante el día, especialmente con alimentos que contengan carbohidratos (almidones). Durante la noche, se coloca una sonda de alimentación a través de la nariz hasta el estómago para brindar un suministro de azúcares o maicena cruda. La sonda se puede colocar en el momento de acostarse y retirarse cada mañana.
Alopurinol, puede disminuir los niveles de ácido úrico en la sangre y reducir el riesgo de gota. Otros medicamentos pueden abarcar aquéllos para la enfermedad renal, lípidos altos y para incrementar las células que combaten la infección.
ENFERMEDAD DE POMPE (GLUCOGENOSIS TIPO II)
Como
en cualquier enfermedad lisosomal el tratamiento se da por un sustitutivo con enzima
recombinante. La alglucosidasa alfa
(myozyme) es una alternativa prometedora y los ensayos clínicos que se están llevando a
cabo en la forma clásica permiten vislumbrar una eficacia en la eliminación del
glucógeno acumulado en corazón, músculo esquelético y sistema nervioso.

REFERENCIAS BIBLIOGRÁFICAS Y NETGRÁFICAS
·
ENFERMEDAD DE
VON-GIERKE – Artículo disponible en el sitio web: http://www.nlm.nih.gov/medlineplus/spanish/ency/article/000338.htm
·
GLUCOGENOSIS
TIPO I – REVISTA CHILENA DE NUTRICIÓN – Artículo dispoble en el sitio web: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0717-75182006000200002
·
GLUCOGENOSIS –
Información disponible en el sitio web: http://www.glucogenosis.net/
·
GLUCOGENOSIS
TIPO II – Información disponible en el sitio web: http://enfermedadpompe.blogspot.com/
·
Werner Muller Estrl,
BIOQUÍMICA – FUNDAMENTOS PARA MEDICINA.- Ed. REVERTÉ - España, S.L.- Barcelona España.- Año 2008. Págs. 528-569
IMÁGEN 1: Pág. 537
IMÁGEN 2: Pág: 541
MUCHAS GRACIAS POR DARTE EL TIEMPO NECESARIO Y LEER ESTA ENTRADA.
QUE TENGAS UN BUEN DÍA
No hay comentarios:
Publicar un comentario